La découverte
 En 1953 et 1954, les docteurs Henry K. Silver et Alex Russell ont décrit séparément des groupes d’enfants petits par rapport à l’âge gestationnel (SGA / Small for Gestational Age ou nés PAG / Petits pour l’Âge Gestationnel) pour lesquels les grossesses ont été compliquées par un retard de croissance intra-utérin (RCIU ou IUGR Intra-Uterine Growth Retardation).
En 1953 et 1954, les docteurs Henry K. Silver et Alex Russell ont décrit séparément des groupes d’enfants petits par rapport à l’âge gestationnel (SGA / Small for Gestational Age ou nés PAG / Petits pour l’Âge Gestationnel) pour lesquels les grossesses ont été compliquées par un retard de croissance intra-utérin (RCIU ou IUGR Intra-Uterine Growth Retardation).
Leurs découvertes communes sont la petite taille sans croissance de rattrapage, le périmètre crânien normal pour l’âge, le visage triangulaire caractéristique, les oreilles implantées plus bas et le petit doigt incurvé. Ces deux groupes de patients sont maintenant considérés comme ayant eu des variations d’une même maladie qu’on appelle aujourd’hui Russell-Silver Syndrome (RSS) en Amérique du Nord et syndrome de Silver-Russell (SRS) en Europe.
Variété des phénotypes
Un aspect intéressant et important du syndrome de Silver-Russell est la variété des phénotypes. Dans ce contexte, un phénotype est l’ensemble de toutes les caractéristiques et anomalies constatées chez un patient qui sont spécifiquement attribuées au SSR/RSS. Certains individus atteints du syndrome de Silver-Russell ont beaucoup de ces traits, donc un phénotype sévère, d’autres ont très peu de ces traits, donc un phénotype léger.
Au départ le SSR/RSS n’était pas considéré comme une maladie génétique car il était rarement récurrent dans une famille et quand c’était le cas, le schéma de transmission ne suivait pas un mode de transmission génétique coutumier. Une meilleure compréhension des transmissions génétiques a amené les scientifiques à conclure que le SSR/RSS est une maladie génétique mais que sa transmission est complexe. Jusqu’à récemment (2005), le SRS/RSS a été associé à des anomalies structurelles du chromosome 7 [DUP7] et à une augmentation potentielle de mutation de fonction d’un gène à empreinte sur le chromosome 7. Cependant, moins de 15 % des cas sont expliqués par les unidisomies maternelles du chromosome 7.
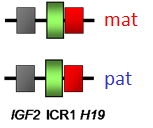
La cause moléculaire la plus fréquente de ce syndrome, et qui concerne au moins 50 % des enfants dits Silver Russell, a été identifiée en 2005 par l’équipe du Pr Le Bouc à l’hôpital Armand Trousseau de Paris. Il s’agit d’un trouble épigénétique (anomalie de méthylation de l’ADN) de la région 11p15 où est situé l’IGF2. L’IGF2 est un facteur de croissance fœtal dont l’expression est donc diminuée en cas d’épimutation de cette région. Cette découverte a donné une grande impulsion pour faciliter le diagnostic difficile de cette maladie et avancer dans la prise en charge thérapeutique des patients concernés.
Anomalies moléculaires retrouvées pour le syndrome de Silver-Russell
- rares anomalies cytogénétiques
- disomie maternelle du chromosome 7 dans environ 7 à 10 % des cas
- perte de méthylation de la Région 11p15 ICR1 dans 50 à 60 % des cas
- environ 30% des SRS/RSS n’ont pas d’anomalie moléculaire identifiée à ce jour.
(D’après Gicquel, Rossignol et al. Nat Genet 2005 Netchine, Rossignol et al. JCEM 2007)